
Adrenoleucodistrofia
 Para imprimir
Para imprimir
Adrenoleucodistrofia ligada al cromosoma X; Adrenomieloneuropatía; Adrenoleucodistrofia cerebral infantil; ALD; Complejo de Schilder-Addison
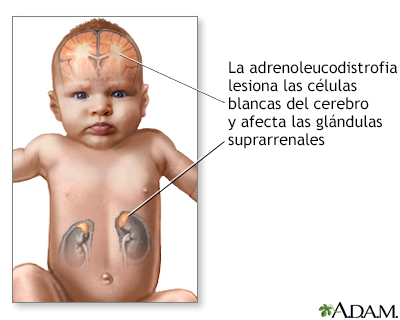

La adrenoleucodistrofia (ADL) describe varios trastornos estrechamente relacionados que interrumpen la descomposición de ciertas grasas. Con frecuencia, estos trastornos se transiten de padres a hijos (hereditarios).
Me gustaría aprender acerca de:
Causas
La ADL por lo regular se trasmite de padres a hijos como un rasgo genético ligado al cromosoma X. Afecta sobre todo a los hombres. Algunas mujeres portadoras pueden tener formas más leves de la enfermedad. Esta enfermedad afecta a alrededor de 1 de cada 20,000 personas de todas las razas.
Algunos casos de ADL se presentan cuando el gen cambia. A esta se le llama espontánea y no es hereditaria.
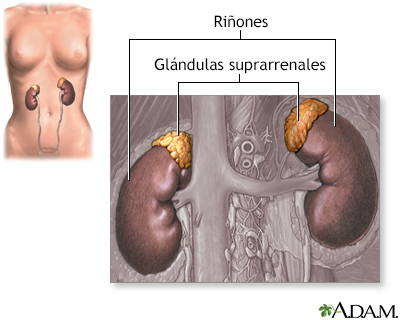
Esta afección ocasiona la acumulación de ácidos grasos de cadena muy larga en el sistema nervioso, en las glándulas suprarrenales y en los testículos. Esto interrumpe la actividad normal en estas partes del cuerpo.
Existen tres categorías principales de esta enfermedad:
- La forma cerebral infantil -- aparece a mediados de la niñez (de 4 a 8 años)
- La adrenomielopatía -- se presenta en hombres de entre 20 años de edad o más tarde en la vida
- Alteración del funcionamiento de las glándulas suprarrenales (llamada enfermedad de Addison o fenotipo similar a Addison) -- la glándula suprarrenal no produce suficientes hormonas esteroides
Síntomas
Los síntomas del tipo cerebral infantil incluyen:
- Cambios en el tono muscular, principalmente espasmos musculares y espasticidad
- Ojos bizcos
- Deterioro de la escritura a mano
- Problemas en la escuela
- Dificultad para entender lo que la gente esta diciendo
- Hipoacusia
- Hiperactividad
- Deterioro del sistema nervioso, incluyendo coma, disminución del control motor fino y parálisis
- Convulsiones
- Dificultad para deglutir
- Deterioro visual o ceguera
Los síntomas de la ADL incluyen:
- Dificultad para controlar la micción
- Posible empeoramiento de la debilidad muscular o rigidez en las piernas
- Problemas con la velocidad de pensamiento y la memoria visual
Los síntomas de la insuficiencia suprarrenal (tipo Addison) incluyen:
- Coma
- Inapetencia
- Aumento del color de la piel
- Pérdida de peso y de masa muscular (desgaste)
- Debilidad muscular
- Vómitos
Pruebas y exámenes
En la mayoría de los estados, se realizan pruebas de ADL a los niños como parte de sus pruebas neonatales de detección. Esto se hace con una punción en el talón para extraer sangre (como parte de las pruebas de detección de rutina en recién nacidos). Las pruebas neonatales no diagnostican la afección, pero pueden ayudar a realizar pruebas y un diagnóstico oportuno.
Los exámenes para esta afección incluyen:
- Niveles sanguíneos de los ácidos grasos de cadena muy larga y hormonas que son producidas por las glándulas suprarrenales
- Estudio cromosómico para buscar cambios en el gen ABCD1 que puede hacer que la persona sea más propensa a desarrollar la enfermedad
- Resonancia magnética de la cabeza
- Biopsia de la piel
- Niveles sanguíneos de los ácidos grasos de cadena muy larga y hormonas que son producidas por las glándulas suprarrenales
Tratamiento
La disfunción suprarrenal se puede tratar con esteroides suplementarios, tales como el cortisol, si la glándula suprarrenal no está produciendo suficientes hormonas.
No existe un tratamiento específico disponible para la ADL ligada al cromosoma X. Un trasplante de médula ósea puede detener el progreso de la afección.
Cuidados de apoyo y observación cuidadosa de la alteración del funcionamiento de la glándula suprarrenal pueden ayudar a mejorar la comodidad y la calidad de vida.
Grupos de apoyo
Los siguientes recursos pueden proporcionar más información sobre la ADL:
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/adrenoleukodystrophy/
- NIH/NLM Genetics Home Reference -- medlineplus.gov/genetics/condition/x-linked-adrenoleukodystrophy/
Expectativas (pronóstico)
La forma infantil de la ADL ligada al cromosoma X es una enfermedad progresiva que lleva a un coma prolongado (estado vegetativo) en aproximadamente 2 años después de la aparición de los síntomas neurológicos. El niño puede vivir en este estado hasta por 10 años hasta que se presenta la muerte.
Las otras formas de esta enfermedad son más leves.
Posibles complicaciones
Pueden presentarse las siguientes complicaciones:
- Crisis suprarrenal
- Estado vegetativo
Cuándo contactar a un profesional médico
Comuníquese con su proveedor de atención médica si:
- Su hijo desarrolla síntomas de adrenoleucodistrofia ligada al cromosoma X
- Su hijo tiene este tipo de adrenoleucodistrofia y está empeorando
Prevención
Se recomienda asesoría genética para parejas que tienen antecedentes familiares de adrenoleucodistrofia ligada al cromosoma X. Las madres de los hijos afectados tienen un 85% de probabilidad de ser portadoras de la afección.
El diagnóstico prenatal de la adrenoleucodistrofia ligada al cromosoma X también está disponible. Se hace mediante una examinación de células de una muestra de vellosidades coriónicas o amniocentesis. Estos exámenes buscan cualquier cambio genético conocido en la familia o niveles de ácidos grasos de cadena muy larga.








Información conexa
| MetabolismoGlándulas suprarre...TestículosGenéticaHiperactividadEstrabismoConvulsionesSustancia blanca d...Deformidad por con...Disminución del es... | Attention deficit ... |
Referencias
James WD, Elston DM, Treat JR, Rosenbach MA, Neuhaus IM. Errors in metabolism. In: James WD, Elston DM, Treat JR, Rosenbach MA, Neuhaus IM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 13th ed. Philadelphia, PA: Elsevier; 2020:chap 26.
Lissauer T, Carroll W. Neurological disorders. In: Lissauer T, Carroll W, eds. Illustrated Textbook of Paediatrics. 6th ed. Philadelphia, PA: Elsevier; 2022:chap 29.
Wangler MF. Defects in metabolism of lipids./Disorders of very-long-chain fatty acids and other peroxisomal functions. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 106.2.